Measurements taken for the purpose of determining employee exposure to benzene are best taken so that the representative average eight-hour exposure may be determined from a single eight-hour sample or two four-hour samples. Short-time interval samples (or grab samples) may also be used to determine average exposure level if a minimum of five measurements are taken in a random manner over the eight-hour work shift. Random sampling means that any portion of the work shift has the same chance of being sampled as any other. The arithmetic average of all such random samples taken on one work shift is an estimate of an employee's average level of exposure for that work shift. Air samples should be taken in the employee's breathing zone (air that would most nearly represent that inhaled by the employee). Sampling and analysis must be performed with procedures meeting the requirements of the standard.
There are a number of methods available for monitoring employee exposures to benzene. The sampling and analysis may be performed by collection of the benzene vapor on charcoal adsorption tubes, with subsequent chemical analysis by gas chromatography. Sampling and analysis may also be performed by portable direct reading instruments, real-time continuous monitoring systems, passive dosimeters or other suitable methods. The employer has the obligation of selecting a monitoring method which meets the accuracy and precision requirements of the standard under his unique field conditions. The standard requires that the method of monitoring must have an accuracy, to a ninety-five percent confidence level, of not less than plus or minus twenty-five percent for concentrations of benzene greater than or equal to 0.5 ppm.
The WISHA laboratory uses NIOSH Method 1500 for evaluation of benzene air concentrations.
(1) WISHA method HYDCB for air samples.
Analyte: Benzene.
Matrix: Air.
Procedure: Adsorption on charcoal, desorption with carbon disulfide, analysis by GC.
Detection limit: 0.04 ppm.
Recommended air volume and sampling rate: 10L at 0.05 to 0.2 L/min.
(a) Principle of the method.
(i) A known volume of air is drawn through a charcoal tube to trap the organic vapors present.
(ii) The charcoal in the tube is transferred to a small, stoppered vial, and the analyte is desorbed with carbon disulfide.
(iii) An aliquot of the desorbed sample is injected into a gas chromatograph.
(iv) The area of the resulting peak is determined and compared with areas obtained from standards.
(b) Advantages and disadvantages of the method.
(i) The sampling device is small, portable, and involves no liquids. Interferences are minimal, and most of those which do occur can be eliminated by altering chromatographic conditions. The samples are analyzed by means of a quick, instrumental method.
(ii) The amount of sample which can be taken is limited by the number of milligrams that the tube will hold before overloading. When the sample value obtained for the backup section of the charcoal tube exceeds twenty-five percent of that found on the front section, the possibility of sample loss exists.
(c) Apparatus.
(i) A calibrated personal sampling pump whose flow can be determined within ± 5 percent at the recommended flow rate.
(ii) Charcoal tubes: Glass with both ends flame sealed, 7 cm long with a 6-mm O.D. and a 4-mm I.D., containing two sections of 20/40 mesh activated charcoal separated by a 2-mm portion of urethane foam. The activated charcoal is prepared from coconut shells and is obtained commercially. The adsorbing section contains 100 mg of charcoal, the back-up section 50 mg. A 3-mm portion of urethane foam is placed between the outlet end of the tube and the back-up section. A plug of silanized glass wool is placed in front of the adsorbing section. The pressure drop across the tube must be less than one inch of mercury at a flow rate of one liter per minute.
(iii) Gas chromatograph equipped with a flame ionization detector.
(iv) Column (10-ft 1/8-in stainless steel) packed with 80/100 Supelcoport coated with twenty percent SP 2100, 0.1 percent CW 1500.
(v) An electronic integrator or some other suitable method for measuring peak area.
(vi) Two-milliliter sample vials with Teflon-lined caps.
(vii) Microliter syringes: 10-microliter 10-uL syringe, and other convenient sizes for making standards, 1-uL syringe for sample injections.
(viii) Pipets: 1.0 mL delivery pipets.
(ix) Volumetric flasks: Convenient sizes for making standard solutions.
(d) Reagents.
(i) Chromatographic quality carbon disulfide (CS2). Most commercially available carbon disulfide contains a trace of benzene which must be removed. It can be removed with the following procedure:
Heat under reflux for two to three hours, 500 mL of carbon disulfide, 10 mL concentrated sulfuric acid, and five drops of concentrated nitric acid. The benzene is converted to nitrobenzene. The carbon disulfide layer is removed, dried with anhydrous sodium sulfate, and distilled. The recovered carbon disulfide should be benzene free. (It has recently been determined that benzene can also be removed by passing the carbon disulfide through 13x molecular sieve.)
(ii) Benzene, reagent grade.
(iii) p-Cymene, reagent grade, (internal standard).
(iv) Desorbing reagent. The desorbing reagent is prepared by adding 0.05 mL of p-Cymene per milliliter of carbon disulfide. (The internal standard offers a convenient means correcting analytical response for slight inconsistencies in the size of sample injections. If the external standard technique is preferred, the internal standard can be eliminated.)
(v) Purified GC grade helium, hydrogen, and air.
(e) Procedure.
(i) Cleaning of equipment. All glassware used for the laboratory analysis should be properly cleaned and free of organics which could interfere in the analysis.
(ii) Calibration of personal pumps. Each pump must be calibrated with a representative charcoal tube in the line.
(iii) Collection and shipping of samples.
(A) Immediately before sampling, break the ends of the tube to provide an opening at least one-half the internal diameter of the tube (2 mm).
(B) The smaller section of the charcoal is used as the backup and should be placed nearest the sampling pump.
(C) The charcoal tube should be placed in a vertical position during sampling to minimize channeling through the charcoal.
(D) Air being sampled should not be passed through any hose or tubing before entering the charcoal tube.
(E) A sample size of ten liters is recommended. Sample at a flow rate of approximately 0.05 to 0.2 liters per minute. The flow rate should be known with an accuracy of at least ± 5 percent.
(F) The charcoal tubes should be capped with the supplied plastic caps immediately after sampling.
(G) Submit at least one blank tube (a charcoal tube subjected to the same handling procedures, without having any air drawn through it) with each set of samples. Take necessary shipping and packing precautions to minimize breakage of samples.
(iv) Analysis of samples.
(A) Preparation of samples. In preparation for analysis, each charcoal tube is scored with a file in front of the first section of charcoal and broken open. The glass wool is removed and discarded. The charcoal in the first (larger) section is transferred to a 2-ml vial. The separating section of foam is removed and discarded; the second section is transferred to another capped vial. These two sections are analyzed separately.
(B) Desorption of samples. Prior to analysis, 1.0 mL of desorbing solution is pipetted into each sample container. The desorbing solution consists of 0.05 uL internal standard per mL of carbon disulfide. The sample vials are capped as soon as the solvent is added. Desorption should be done for thirty minutes with occasional shaking.
(C) GC conditions. Typical operating conditions for the gas chromatograph are:
(I) 30 mL/min (60 psig) helium carrier gas flow.
(II) 30 mL/min (40 psig) hydrogen gas flow to detector.
(III) 240 mL/min (40 psig) air flow to detector.
(IV) 150°C injector temperature.
(V) 250°C detector temperature.
(VI) 100°C column temperature.
(D) Injection size. 1 µL.
(E) Measurement of area. The peak areas are measured by an electronic integrator or some other suitable form of area measurement.
(F) An internal standard procedure is used. The integrator is calibrated to report results in ppm for a ten liter air sample after correction for desorption efficiency.
(v) Determination of desorption efficiency.
(A) Importance of determination. The desorption efficiency of a particular compound can vary from one laboratory to another and from one lot of chemical to another. Thus, it is necessary to determine, at least once, the percentage of the specific compound that is removed in the desorption process, provided the same batch of charcoal is used.
(B) Procedure for determining desorption efficiency. The reference portion of the charcoal tube is removed. To the remaining portion, amounts representing 0.5X, 1X, and 2X and (X represents target concentration) based on a 10 L air sample are injected into several tubes at each level. Dilutions of benzene with carbon disulfide are made to allow injection of measurable quantities. These tubes are then allowed to equilibrate at least overnight. Following equilibration they are analyzed following the same procedure as the samples. Desorption efficiency is determined by dividing the amount of benzene found by amount spiked on the tube.
(f) Calibration and standards. A series of standards varying in concentration over the range of interest is prepared and analyzed under the same GC conditions that will be used on the samples. A calibration curve is prepared by plotting concentration (mg/mL) versus peak area.
(g) Calculations. Benzene air concentration can be calculated from the following equation:
mg/m3=(A)(B)/(C)(D)
Where: A=µg/mL benzene, obtained from the calibration curve
B=desorption volume (1 mL)
C=Liters of air sampled
D=desorption efficiency
The concentration in mg/m3 can be converted to ppm (at 25° C and 760 mm) with the following equation:
ppm=(mg/m3)(24.46)/(78.11)
Where: 24.46=molar volume of an ideal gas
25° C and 760 mm
78.11=molecular weight of benzene
(h) Backup data.
(i) Detection limit-air samples.
The detection limit for the analytical procedure is 1.28 mg with a coefficient of variation of 0.023 at this level. This would be equivalent to an air concentration of 0.04 ppm for a 10 L air sample. This amount provided a chromatographic peak that could be identifiable in the presence of possible interferences. The detection limit data were obtained by making 1 µL injections of a 1.283 µg/mL standard.
Injection | Area Count | |
1. . . . 2. . . . 3. . . . 4. . . . 5. . . . 6. . . . | 655.4 617.5 662.0 641.1 636.4 629.2 | 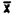 = 640.2 = 640.2SD = 14.9 CV = 0.023 |
(ii) Pooled coefficient of variation-Air Samples. The pooled coefficient of variation for the analytical procedure was determined by 1 uL replicate injections of analytical standards. The standards were 16.04, 32.08, and 64.16 ug/mL, which are equivalent to 0.5, 1.0, and 2.0 ppm for a 10 L air sample respectively.
Area Counts | |||
Injection | 0.5 ppm | 1.0 ppm | 2.0 ppm |
1. . . . 2. . . . 3. . . . 4. . . . 5. . . . 6. . . . 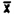 = =SD = CV = | 3996.5 4059.4 4052.0 4027.2 4046.8 4137.9 4053.3 47.2 0.0116 | 8130.2 8235.6 8307.9 8263.2 8291.1 8288.8 8254.0 62.5 0.0076 | 16481 16493 16535 16609 16552 16618 16548.3 57.1 0.0034 |
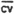 = 0.008. . . . = 0.008. . . . | . . . . | . . . . | . . . . |
(iii) Storage data-air samples.
Samples were generated at 1.03 ppm benzene at eighty percent relative humidity, 22° C, and 643 mm. All samples were taken for fifty minutes at 0.2 L/min. Six samples were analyzed immediately and the rest of the samples were divided into two groups by fifteen samples each. One group was stored at refrigerated temperature of -25° C, and the other group was stored at ambient temperature (approximately 23° C). These samples were analyzed over a period of fifteen days. The results are tabulated below.
percent recovery
Day Analyzed | Refrigerated | Ambient | ||||
0. . . . | 97.4 | 98.7 | 98.9 | 97.4 | 98.7 | 98.9 |
0. . . . | 97.1 | 100.6 | 100.9 | 97.1 | 100.6 | 100.9 |
2. . . . | 95.8 | 96.4 | 95.4 | 95.4 | 96.6 | 96.9 |
5. . . . | 93.9 | 93.7 | 92.4 | 92.4 | 94.3 | 94.1 |
9. . . . | 93.6 | 95.5 | 94.6 | 95.2 | 95.6 | 96.6 |
13. . . . | 94.3 | 95.3 | 93.7 | 91.0 | 95.0 | 94.6 |
15. . . . | 96.8 | 95.8 | 94.2 | 92.9 | 96.3 | 95.9 |
(iv) Desorption data.
Samples were prepared by injecting liquid benzene onto the A section of charcoal tubes. Samples were prepared that would be equivalent to 0.5, 1.0, and 2.0 ppm for a 10 L air sample.
Sample | 0.5 ppm | 1.0 ppm | 2.0 ppm |
1. . . . | 99.4 | 98.8 | 99.5 |
2. . . . | 99.5 | 98.7 | 99.7 |
3. . . . | 99.2 | 98.6 | 99.8 |
4. . . . | 99.4 | 99.1 | 100.0 |
5. . . . | 99.2 | 99.0 | 99.7 |
6. . . . | 99.8 | 99.1 | 99.9 |
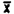 = = | 99.4 | 98.9 | 99.8 |
SD = | 0.22 | 0.21 | 0.18 |
CV = | 0.0022 | 0.0021 | 0.0018 |
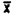 = 99.4 = 99.4 |
(v) Carbon disulfide.
Carbon disulfide from a number of sources was analyzed for benzene contamination. The results are given in the following table. The benzene contaminant can be removed with the procedures given in (d)(i) of this subsection.
sample | ug Benzene/mL | ppm equivalent (for 10 L air sample) |
Aldrich Lot 83017. . . . | 4.20 | 0.13 |
Baker Lot 720364. . . . | 1.0† | 0.03 |
Baker Lot 822351. . . . | 1.0† | 0.03 |
Malinkrodt Lot WEMP. . . . | 1.74 | 0.05 |
Malinkrodt Lot WHGA. . . . | 5.65 | 0.18 |
Treated CS2. . . . | 2.90 | 0.09 |
(2) WISHA laboratory method for bulk samples.
Analyte: Benzene.
Matrix: Bulk samples.
Procedure: Bulk samples are analyzed directly by high performance liquid chromatography (HPLC) or by capillary gas chromatography. See laboratory manual for GC procedure.
Detection limits: 0.01% by volume.
(a) Principle of the method.
(i) An aliquot of the bulk sample to be analyzed is injected into a liquid chromatograph or gas chromatograph.
(ii) The peak area for benzene is determined and compared to areas obtained from standards.
(b) Advantages and disadvantages of the method.
(i) The analytical procedure is quick, sensitive, and reproducible.
(ii) Reanalysis of samples is possible.
(iii) Interferences can be circumvented by proper selection of HPLC parameters or GC parameters.
(iv) Samples must be free of any particulates that may clog the capillary tubing in the liquid chromatograph. This may require distilling the sample or clarifying with a clarification kit.
(c) Apparatus.
(i) Liquid chromatograph equipped with a UV detector or capillary gas chromatograph with FID detector.
(ii) HPLC column that will separate benzene from other components in the bulk sample being analyzed. The column used for validation studies was a Waters uBondapack C18, 30 cm x 3.9 mm.
(iii) A clarification kit to remove any particulates in the bulk if necessary.
(iv) A micro-distillation apparatus to distill any samples if necessary.
(v) An electronic integrator or some other suitable method of measuring peak areas.
(vi) Microliter syringes-10 uL syringe and other convenient sizes for making standards. 10 uL syringe for sample injections.
(vii) Volumetric flasks, 5 mL and other convenient sizes for preparing standards and making dilutions.
(d) Reagents.
(i) Benzene, reagent grade.
(ii) HPLC grade water, methyl alcohol, and isopropyl alcohol.
(e) Collection and shipment of samples.
(i) Samples should be transported in glass containers with Teflon-lined caps.
(ii) Samples should not be put in the same container used for air samples.
(f) Analysis of samples.
(i) Sample preparation.
If necessary, the samples are distilled or clarified. Samples are analyzed undiluted. If the benzene concentration is out of the working range, suitable dilutions are made with isopropyl alcohol.
(ii) HPLC conditions.
The typical operating conditions for the high performance liquid chromatograph are:
(A) Mobile phase-Methyl alcohol/water, 50/50.
(B) Analytical wavelength-254 nm.
(C) Injection size-10 µL.
(iii) Measurement of peak area and calibration.
Peak areas are measured by an integrator or other suitable means. The integrator is calibrated to report results % in benzene by volume.
(g) Calculations.
Since the integrator is programmed to report results in % benzene by volume in an undiluted sample, the following equation is used:
% Benzene by Volume = A x B
Where: A = % by volume on report
B = Dilution Factor
(B = 1 for undiluted sample)
(h) Backup data.
(i) Detection limit-bulk samples.
The detection limit for the analytical procedure for bulk samples is 0.88 ug, with a coefficient or variation of 0.019 at this level. This amount provided a chromatographic peak that could be identifiable in the presence of possible interferences. The detection limit data were obtained by making 10 uL injections of a 0.10% by volume standard.
1. . . . | 45386 | |
2. . . . | 44214 | |
3. . . . | 43822 | 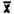 = 44040.1 = 44040.1 |
4. . . . | 44062 | SD = 852.5 |
6. . . . | 42724 | CV = 0.019 |
(ii) Pooled coefficient of variation-bulk samples.
The pooled coefficient of variation for analytical procedure was determined by 50 uL replicate injections of analytical standards. The standards were 0.01, 0.02, 0.04, 0.10, 1.0, and 2.0% benzene by volume.
Injection No. | 0.01 | 0.02 | 0.04 | 0.10 | 1.0 | 2.0 |
1. . . . | 45386 | 84737 | 166097 | 448497 | 4395380 | 9339150 |
2. . . . | 44241 | 84300 | 170832 | 441299 | 4590800 | 9484900 |
3. . . . | 43822 | 83835 | 164160 | 443719 | 4593200 | 9557580 |
4. . . . | 44062 | 84381 | 164445 | 444842 | 4642350 | 9677060 |
5. . . . | 44006 | 83012 | 168398 | 442564 | 4646430 | 9766240 |
6. . . . | 42724 | 81957 | 173002 | 443975 | 4646260 | . . . . |
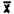 = = | 44040.1 | 83703.6 | 167872 | 444149 | 4585767 | 9564986 |
SD = | 852.5 | 1042.2 | 3589.8 | 2459.1 | 96839.3 | 166233 |
CV = | 0.0194 | 0.0125 | 0.0213 | 0.0055 | 0.0211 | 0.0174 |
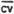 = = | 0.017 |